Automated Umbrella Sampling Simulation
 sampling simulation automated by **CHAPERON*****g***.")
Carry out GROMACS-based enhanced (umbrella) sampling simulation using the automated workflow of CHAPERONg
1. Umbrella Sampling: Introduction
The accurate calcualtion of the binding energy of protein complexes is both a challenge and an important goal in computational biomolecular study and drug design. Umbrella sampling, an enhanced sampling technique for molecular dynamics simulation, is one of the widely-used methods employed in such free energy calculations as well as to explore the dissociation process of protein-ligand systems. It involves the pulling of apart of a protein complexed with, e.g., a small-molecule ligand, or another protein monomer in a multimeric protein over several overlapping simulation windows. The pulling simulation is facilitated by a biasing potential along a reaction coordinate. Each window is subjected to an unbiased MD simulation. The sampled windows are then combined using a method such as the weighted histogram analysis method (WHAM) which is implemented in GROMACS. The WHAM method is used to calculate the potential of mean force (PMF), and hence, the free energy of binding (Kästner, 2011).
2. Choosing the Type of Simulation and System
As shown in the Figure below, CHAPERONg enables the automated enhanced (umbrella) sampling simulation with GROMACS for protein complex systems, including:
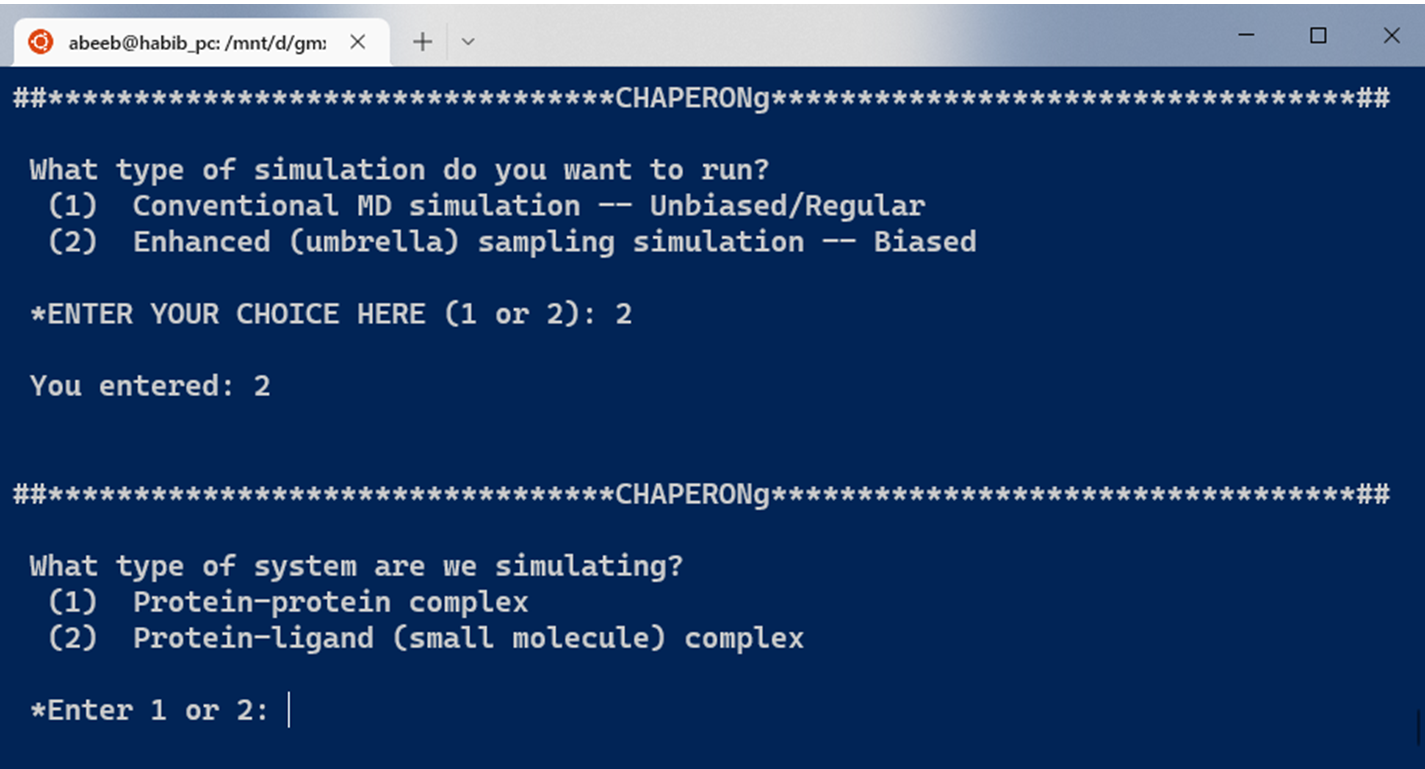
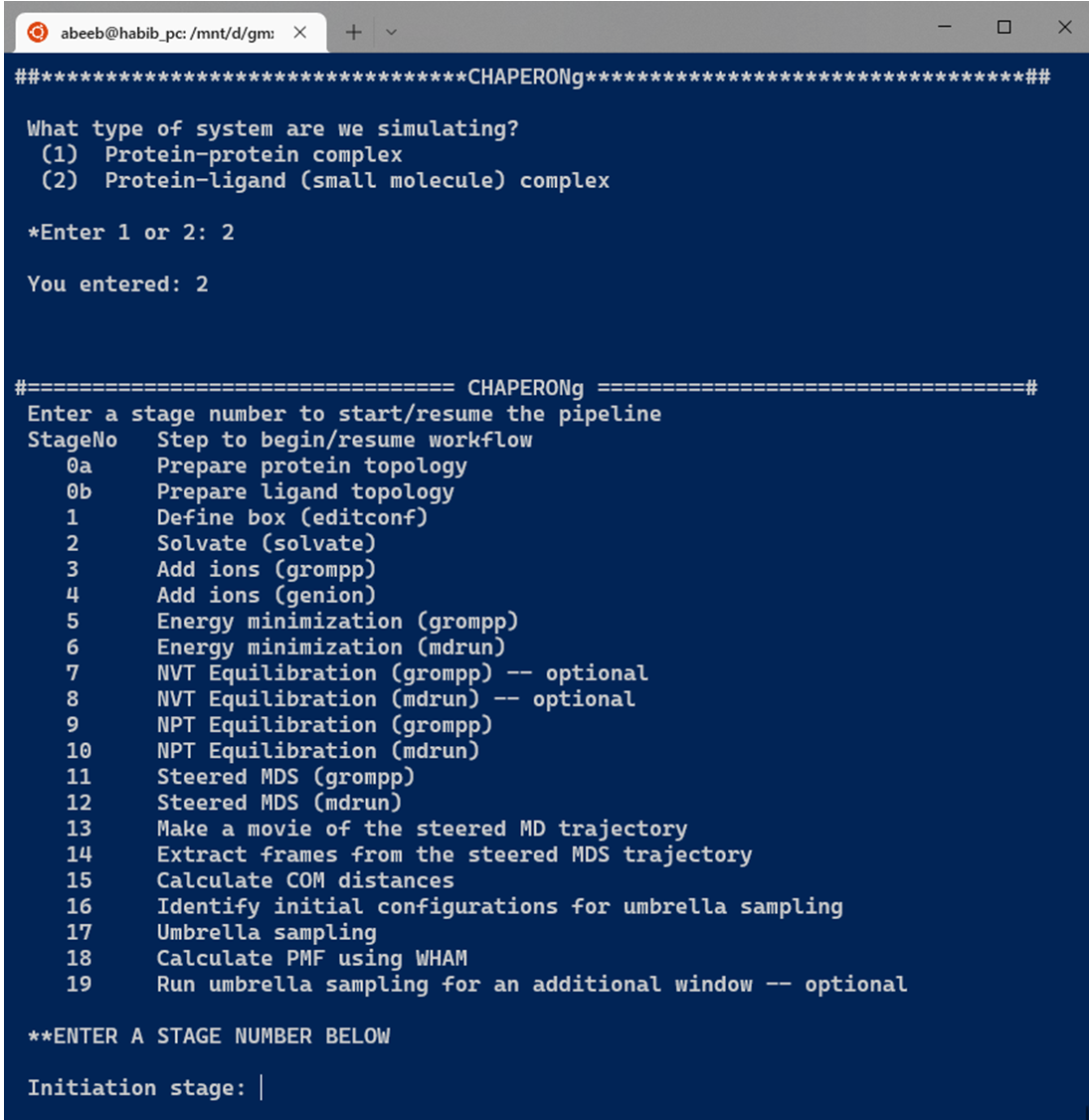
The initial steps involved in the umbrella sampling simulation of these systems as facilitated by CHAPERONg are broadly similar to those described for the conventional, unbiased MD simulation in the previous parts of these documentation. The umbrella sampling simulation, however, features additional steps as shown in the snapshot below. For protein-protein complexes, the simulation steps have been divided into 19 key steps in CHAPERONg, with an additional step of ligand topology preparation in the case of a protein-small-molecule ligand system (see snapshot below).
3. General Simulation Steps
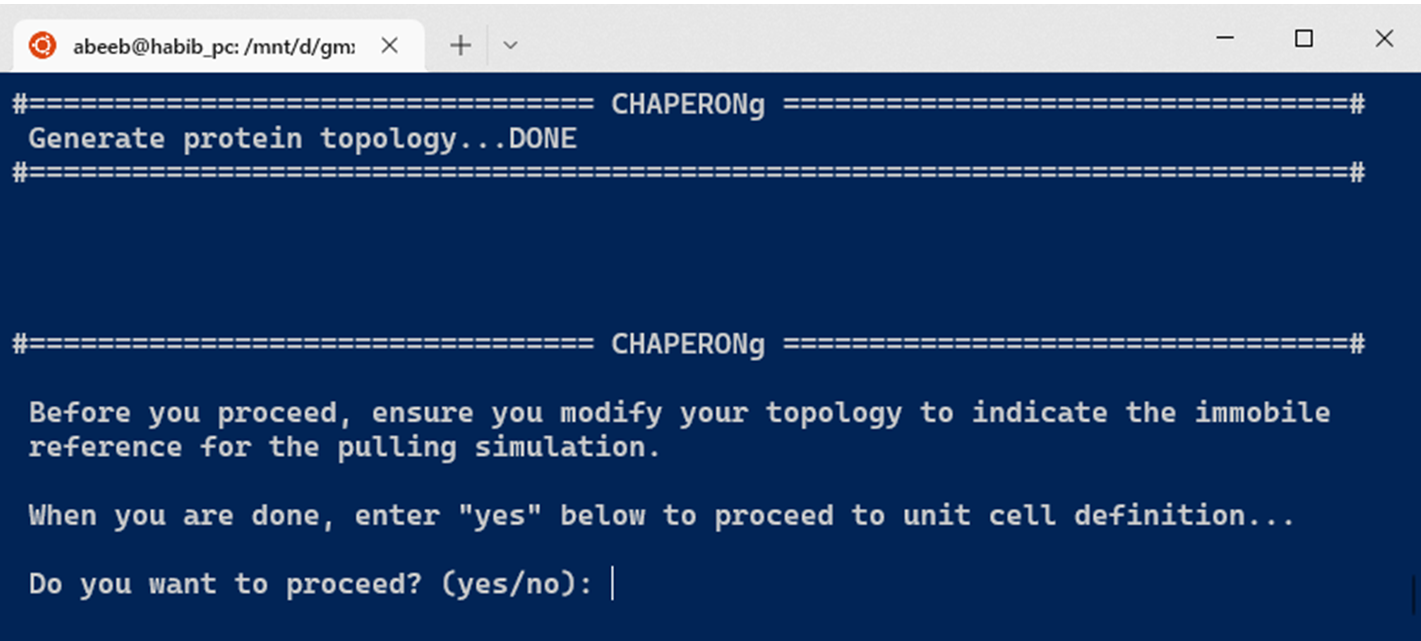
The initial steps of the umbrella sampling simulation (as shown above) are similar to those of the conventional MD simulation previously described. The simulation begins with the preparation of protein topology, after which the user is prompted to confirm that the reference group for the steered MD has been indicated in the topology file if required (see snapshot below).
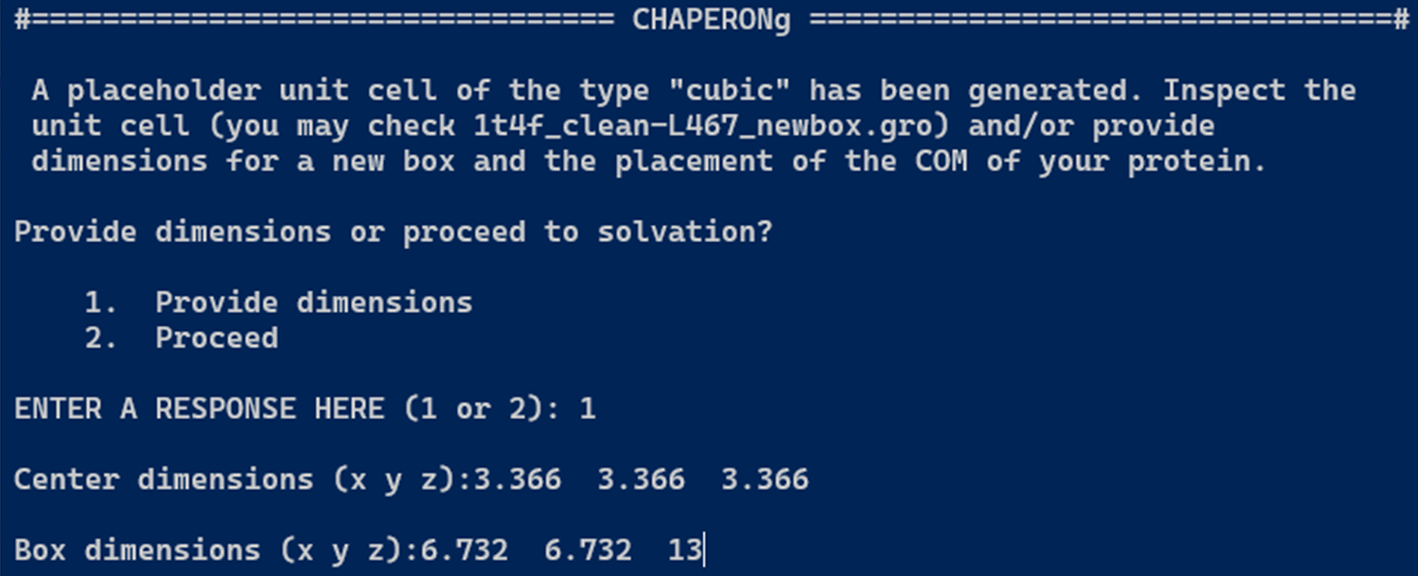
In the case of a protein-small-molecule complex, the topology preparation for the ligand and the complex step is included before the box definition, solvation, adding ions, energy minimization, and equilibration steps. At the box definition step, a placeholder cubic unit cell is first generated. The user is then prompted to inspect the unit cell parameters and provide the new coordinates to use for the center and the box vectors.
4. Steered MD Simulation
The gmx grompp module requires an input .mdp parameter file to generate the appropriate .tpr file needed by gmx mdrun module to run the pulling simulation. Two output files are written, pullf.xvg (which contains the record of the pull force over time) and pullx.xvg (which contains the displacement of the pulled groups with time). Additionally, a plot of the force against the displacement is generated and, together with the other files, saved in a folder named steered_MD.
5. Making a Movie of the Steered MD Simulation
To make a movie of the pulling simulation trajectory, the total number of frames in the trajectory can be summarized into a smaller number of frames extracted at regular intervals. This number can be specified using either the --movieFrame at the launch of CHAPERONg, or provided in the parameter file paraFile.par. Note that this same parameter is set for making a movie for unbiased MD simulation trajectory in the case of the conventional MD simulation as described earlier, and the procedures involved are similar. If the movieFrame parameter is not provided, the user is prompted to indicate whether a default 200-frame movie should be made or a different number of frames is desired. CHAPERONg uses PyMOL to create snapshots of the frames.
6. Extract Coordinates from the Steered MD Trajectory
Using the gmx trjconv module, the frames in the trajectory are extracted and saved as individual coordinates into the folder coordinates_SMD. If a folder from a previous run exists in the working directory, it is backed up before the folder for a new run is created.
7. Calculate the Center-of-Mass Distance for each frame
For each coordinate in the coordinates_SMD folder (the extracted structures), the distance between the specified groups that define the reaction coordinate are calculated using the gmx distance module. For each of the frames (i.e. structures), a temporary time-distance file is written. The distance files are then used to generate the summary file distances_summary.txt, which is a list of the COM distances with time for the different frames .
8. Identify Initial Configurations for Umbrella Sampling
To identify the starting configurations to use for the umbrella sampling windows, the spacing of the windows needs to be defined using the parameter us_window_spacing in the input parameter file paraFile.par. This parameter is read by the CHAP_set_US_starting_configs.py script which then identifies the frames that correspond to the specified spacing. These frames are written to the file configurations_list.txt and used as the starting configurations for the umbrella sampling windows.
9. Umbrella Sampling
Next, umbrella sampling is run for each of the windows. The simulation is prepared by first running a short NPT equilibration for each window. This step requires an appropriate .mdp parameter file, that should be named as npt_umbrella.mdp, which is needed by the gmx grompp module. This is followed by the umbrella sampling and an .mdp file named md_umbrella.mdp is required. During umbrella sampling, a pullf.xvg file (which records the progression of force with time) and a pullx.xvg file (which records the displacement with time) are generated for each of the sampling window. In addition, three important files are produced, viz:
-
tpr_files.dat: a files that lists the names of all the .tpr files generated.
-
pullf_files.dat: a files that lists the names of all the pullf.xvg files generated.
-
pullx_files.dat: a files that lists the names of all the pullx.xvg files generated.
The tpr_files.dat and pullf_files.dat files will be passed to the subsequent data analysis.
10. Calculate the PMF using the WHAM Algorithm
The potential of mean force (PMF) is then computed from the analysis of the output data generated by the umbrella sampling simulations. The weighted histogram analysis method (WHAM), available in GROMACS via the gmx wham module, is used to achieve the reconstruction of the PMF curve. This step produces the PMF_profile.xvg and the umbrella_sampling_histograms.xvg files. In addition, a second plot of the PMF curve with the energy minimum adjusted to zero, PMF_profile_YminAdjusted.xvg, is produced along with the .PNG images of all the plots. Finally, the binding free energy $ {\Delta}G$ is calculated by taking the difference between the lowest and the highest values of the PMF curve. This calculated value is written to a file named summary_dG.dat. All the output files from the PMF calculations are saved in the folder Data_Analysis_PMF.
11. Running Umbrella Sampling for Additional Windows
CHAPERONg provides an option to run umbrella sampling for additional windows in case the histograms from a previous run show insufficient sampling at certain windows. With the option, the user is prompted to provide the frame number of the SMD coordinate (from the previous pulling simulation) to use as the starting configuration for the additional umbrella sampling window. The umbrella sampling for the specified window is then run as described above. This cycle can be repeated
.
Related: BioMoDes: Conformational Ensemble & Dynamics Analysis Tools | CHAPERONg Tutorials
I try my best to make the information on this website as accurate as possiblef you find any errors in the contents of this page or any other page on this website, I would greatly appreciate that you kindly get in touch with me at contact@abeebyekeen.com. Also, you are welcome to reach out for assistance and collaboration.