Learn about what CHAPERONg can do and how it does it.
After you’ve downloaded and installed CHAPERONg (you can find how to do so here), follow the explanations below to know what it can do and understand how it runs.
The run_CHAPERONg executable can be called from any directory (i.e. the working directory) after installation of the CHAPERONg code. The input parameters for running CHAPERONg can be provided using one or both of two ways:
command-line options, or
input parameter fileparaFile.par.
Command-line options (click the 'Code' drop-down below to reveal the contents)
(<int> == integer; <str> == string)
Required parameter
####################################################################################
-i, --input <str> Input coordinate file (.pdb or .gro)
Optional parameters
####################################################################################
-h, --help Print the shorter version of this help
-b, --bt <str> Box type: cubic (default), dodecahedron, triclinic, etc.
-T, --nt <int> Number of threads to use [default: 0 (gmx guesses)]
-g, --nb gpu Calculate non-bonded interactions on gpu
-G, --gpu_id <str> List ID(s) of unique GPU devices available for use
-p, --deffnm <str> Set filename prefix (default for outputs: "md_filename")
-a, --auto_mode Automation mode [options: full, semi(default)]. full: Use
default parameters & do common analyses (less prompts)
-H, --Help Print more, advanced options
-s, --water <str> Water model: tip3p, spc, spce, etc. (ff-dependent)
-f, --ff <str> Force-field: charmm27, amber94, oplsaa, gromos54a7, etc.
(Enter "wd" if forcefield is in working directory)
-P, --posname <str> Name of the positive ion (default: NA)
-N, --negname <str> Name of the negative ion (default: CL)
-c, --conc <int> Set salt concentration (mol/L) for the system
-W, --maxwarn <int> Number of allowed warnings (default is 0)
-M, --mmgpath <str> Absolute path to gmx binary to use for g_mmpbsa
-E, --gmx_exe <str> Path to gmx to use for all gmx runs except g_mmpbsa
(Default is to use the gmx set in the environment)
-v, --version Print the installed version of CHAPERONg
-t, --temp <int> Simulation temperature in kelvin
--ntmpi <int> Number of thread-MPI ranks [default: 0 (gmx guesses)]
--ntomp <int> Number of OpenMP threads per MPI rank; default: 0 (guess)
--paraFile <str> Name of the CHAPERONg input parameter file
--inputtraj <str> Corrected trajectory to generate and use for analyses
(options: noPBC, nojump, center, fit, combo)
--clustr_cut <float> RMSD cut-off (nm) for cluster membership (default: 1.0)
--clustr_methd <str> Method for cluster determination: gromos (default),
linkage, jarvis-patrick, monte-carlo, diagonalization
--frame_beginT <int> Time (ps) of first frame to read from trajectory
--frame_endT <int> Time (ps) of last frame to read from trajectory
--dt <int> Interval (ps) at which frames are taken from trajectory
--mmFrame <int> Number of frames to be extracted for g_mmpbsa
--movieFrame <int> Number of frames to extract and use for movie
--trFrac <int> Fraction of trajectory to use for g_mmpbsa
(enter 1 for all, 2 for 2nd half, 3 for last 3rd, etc.)
--kde_opt <int> Range (above and below the estimate) to test for the
optimization of histogram number of bins for KDE
--path_av_plot<str> Path to input files for average of replica plots
--dist <float> Solute-box distance (distance to box edge; default: 1.0)
--bg Run production mdrun in the background with "nohup"
--ter <prompt> Interactively choose the N- & C-termini protonation
states (default: ionized with NH3+ & COO-)
paraFile.par (click the 'Code' drop-down below to reveal the contents)
; this is an input parameter file for CHAPERONg
; lines beginning with ";" are comments
; lines beginning with "#" are disabled/unset parameters
; delete the "#" to activate and set a parameter
#################################################################
; GENERAL PARAMETERS
; Input coordinate file (.pdb or .gro)
# input = x
; Simulation box type: cubic, dodecahedron, triclinic, octahedron
# bt = x
; Number of threads to use
# nt = x
; Use system GPU for non-bonded interactions
# nb = gpu
; List ID(s) of unique GPU device(s) available for use
# gpu_id = x
; Automation mode: full, semi
# auto_mode = full
; Define filename prefix
# deffnm = x
; Water model i.e. tip3p, spc
# water = tip3p
; Force-field (e.g., charmm27, amber94, amber99sb, gromos54a7, oplsaa, etc.),
; full list at https://manual.gromacs.org/current/user-guide/force-fields.html
; use "wd" if force-field is in working directory
# ff = wd
; Number of thread-MPI ranks
# ntmpi = x
; Number of OpenMP threads per MPI rank
# ntomp = x
; Number of frames to extract and use for movie
# movieFrame = 200
; Name of the positive ion
# posname = NA
; Name of the negative ion
# negname = CL
; Salt concentration (mol/L)
# conc = 0.1
; Simulation temperature (in kelvin)
# temp = 300
; Number of allowed warnings
# maxwarn = x
; Solute-box distance i.e. distance to box edge
# dist = 1.0
; Corrected trajectory to use for analyses (options: noPBC,
; nojump, fit, combo)
# inputtraj = combo
; Absolute path to the gmx executable to use for
; all gmx runs except those involving mmpbsa
# gmx_exe = x
; Parameter for umbrella sampling window spacing
# us_window_spacing = 0.2
; PARAMETERS FOR g_mmpbsa CALCULATIONS
; Absolute path to gmx binary to be used for mmpbsa
# mmgpath = /home/abeeb/local/gromacs5.1/bin/gmx
; Fraction of trajectory to use for g_mmpbsa calculations
# trFrac = x
; Number of frames to extract for g_mmpbsa
# mmFrame = 400
; Time (in ps) to begin reading frames from trajectory for g_mmpbsa
# mmBegin = x
; PARAMETERS FOR CLUSTERING
; RMSD cut-off (nm) for two structures to be neighbors
# clustr_cut = 0.15
; Method to use for cluster determination
# clustr_methd = gromos
; Time (ps) of first frame to read from trajectory for clustering
# frame_beginT = 0
; Time (ps) of last frame to read from trajectory for clustering
# frame_endT = 0
; Time interval (ps) at which frame is taken from the trajectory
# dt = 1
; PARAMETERS FOR AVERAGED PLOT OF REPLICA ANALYSIS PLOTS
; Path to the directory containing input replica plots
# path_av_plot = x
; Data label for the input replica plots (e.g. RMSD, Rg, RMSF, SASA, etc.)
# data_label = x
2. Automation modes of CHAPERONg
CHAPERONg can be run in two modes of automation which can be set using the flag auto_mode or a:
Fully automated mode: This mode is set with the --auto_mode full or -a full flag. The simulation steps and post-simulation analyses are all run automatically based on the type of simulation and user-provided parameters. Running CHAPERONg in this mode is recommended as it only presents a few GROMACS prompts which occur in situations where CHAPERONg would not be able to make a decision without an input from the user.
Semi-automated mode: This is the default mode when the --auto_mode or -a flag is not provided, or set to --auto_mode semi or -a semi by the user at the launch of the program. Most steps are still automated, but the user is prompted to enter some input when there is a choice to be made while running GROMACS.
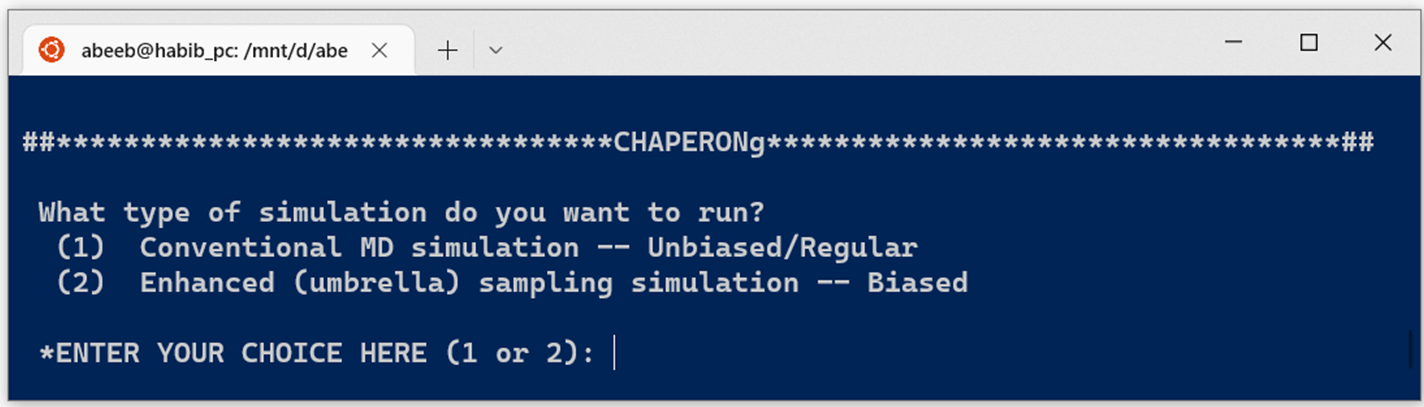
3. Supported simulation types and systems
The current version of CHAPERONg supports and automates the following simulation types:
Conventional/regular (unbiased) MD simulation
Enhanced (umbrella) sampling simulation
GROMACS MD simulation types supported by CHAPERONg
I try my best to make the information on this website is as accurate as possible, but if you find any errors in the contents of this page or any other page on this website, I would greatly appreciate that you kindly get in touch with me at contact@abeebyekeen.com. Also, you are welcome to reach out for assistance and collaboration.